Key points
- Creutzfeldt-Jakob disease (CJD) is a rapidly progressive and always fatal neurodegenerative disorder.
- It is believed to be caused by prions, which can cause abnormal folding of normal proteins in the brain.
- CJD occurs worldwide and the estimated annual incidence in many countries, including the United States, has been reported to be about 1-2 cases per 1 million population.
- CDC supports the National Prion Disease Pathology Surveillance Center at Case Western Reserve University for state-of-the-art diagnostic testing.
Overview
CJD was first recognized in the 1920s and is classified as a transmissible spongiform encephalopathy along with other prion diseases that occur in humans and animals. The risk of CJD increases with a person's age, but overall, the risk for CJD is very low, with an estimated annual incidence in many countries of about 1 to 2 cases per 1 million people
In about 85% of patients, CJD occurs as a sporadic disease with no recognizable pattern of transmission. A smaller proportion of patients (5-15%) develop familial CJD due to inherited mutations in the prion protein gene. Less than 1% of CJD cases are classified as iatrogenic, resulting from contact with prions in a healthcare setting or from biological products.
Confirming CJD in a patient requires neuropathologic and/or immunodiagnostic testing of brain tissue obtained either by biopsy or autopsy.
Clinical features
CJD typically affects people 55 years old and older. The primary symptom is dementia with early neurologic signs that may include trouble walking, sudden jerky movements, and visual disturbances.
Once symptoms begin, the patient's decline is rapid, with a median of 4-5 months until death.
Diagnosis
There are standard diagnostic criteria for definite, probable and possible sporadic CJD cases. There are separate diagnostic criteria for cases that may be iatrogenic and those that may be familial.
Sporadic CJD
Definite
Diagnostic criteria include:
- Standard neuropathological techniques
AND/OR
- Immunocytochemically
AND/OR
- Western blot confirmed protease-resistant PrP
AND/OR
- Presence of scrapie-associated fibrils
Probable
Diagnostic criteria include:
- Neuropsychiatric disorder
- Plus positive RT-QuIC in cerebrospinal fluid (CSF) or other tissues
OR
Rapidly progressive dementia and at least 2 out of these 4 clinical features:
- Myoclonus
- Visual or cerebellar signs
- Pyramidal/extrapyramidal signs
- Akinteic mutism
AND
A positive result on at least 1 of the following laboratory tests:
- a typical EEG (periodic sharp wave complexes) during an illness of any duration
- a positive 14-3-3 CSF assay in patients with a disease duration of less than 2 years
- High signal in caudate/putamen on magnetic resonance imaging (MRI) brain scan or at least two cortical regions (temporal, parietal, occipital) either on diffusion-weighted imaging (DWI) or fluid attenuated inversion recovery (FLAIR)
AND
- Without routine investigations indicating an alternative diagnosis
Possible
Progressive dementia; and at least 2 out of these 4 clinical features:
- Myoclonus
- Visual or cerebellar signs
- Pyramidal/extrapyramidal signs
- Akinteic mutism
AND
The absence of a positive result for any of the 4 tests above that would classify a case as "probable"
AND
Duration of illness less than 2 years
AND
Without routine investigations indicating an alternative diagnosis
Iatrogenic CJD
Diagnostic criteria include:
- Progressive cerebellar syndrome in a recipient of human cadaveric-derived pituitary hormone
OR
- Sporadic CJD with a recognized exposure risk, such as neurosurgery with dura mater implantation
Familial CJD
Diagnostic criteria include:
- Definite or probable CJD and definite or probable CJD in a first-degree relative
AND/OR
- Neuropsychiatric disorder and disease-specific PrP gene mutation
Patient management
Treatment of prion diseases involves supportive care. There is unfortunately no specific therapy that has been shown to stop the progression of these diseases.
Death rates
Creutzfeldt-Jakob disease deaths and age-adjusted death rate, United States, 1979–2022*
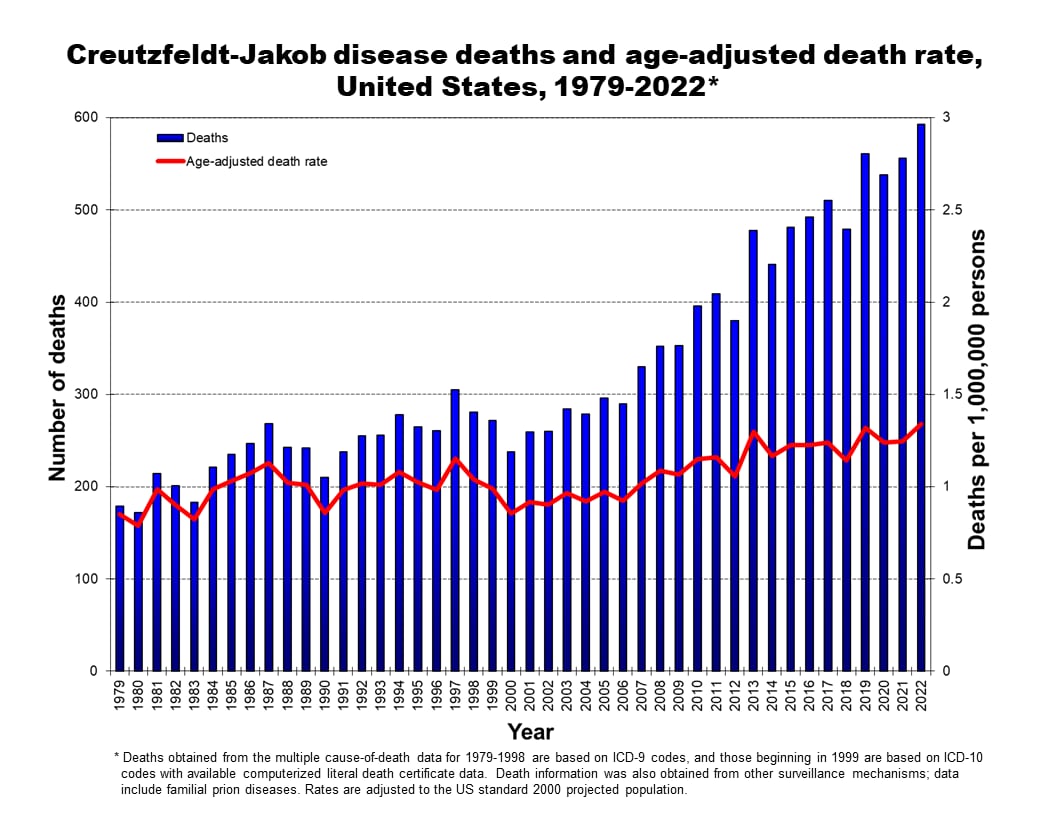
*Deaths obtained from multiple cause-of-death data include all forms of human prion disease and are based on ICD-9 and ICD-10 codes and available computerized literal death certificate data. Some modifications have been made to these data based on relevant information obtained from other surveillance mechanisms. Rates are adjusted to the US standard 2000 projected population.
Not everyone who is thought to have died from prion disease has had their brain tissue analyzed, which is required to confirm prion disease. Because of this, these case counts include people with CJD based on clinical tests if a prion disease diagnosis is listed on their death certificate. A recently developed clinical laboratory test has been shown to strongly indicate prion disease when positive.
Annual numbers of people with this positive test but no brain tissue analyses can be found on the National Prion Disease Pathology Surveillance Center website (NPDPSC Tables). More information on the test is available here.
Similar diseases
It's important to note that classic CJD is different than variant CJD (vCJD). vCJD is a prion disease caused by eating cow products from cattle infected with Bovine Spongiform Encephalopathy (BSE). CJD is not related to BSE.
Resources
- University of California, San Francisco: Memory and Aging Center Creutzfeldt-Jakob Disease Website
- The UK Creutzfeldt-Jakob Disease Surveillance Unit University of Edinburgh, Scotland
- Prion disease incidence in the United State, 2003-2015 Neurology® 2020;94:1-5.
- Diagnostic and prognostic value of human prion detection in cerebrospinal fluid.Ann Neurol. 2017 Jan;81(1):79-92. doi: 10.1002/ana.24833.
- Creutzfeldt-Jakob Disease Surveillance and Diagnosis Clinical Infectious Diseases. 2005; 41:834–836.
- Creutzfeldt-Jakob Disease Not Related to a Common Venue – New Jersey, 1995 – 2004, MMWR. 2004;53(Early Release):1-4.
- Creutzfeldt-Jakob Disease in Unusually Young Patients Who Consumed Venison. Archives of Neurology, 2001;58:1673-1678.
- Creutzfeldt-Jakob Disease in the United States: 1979-1998. Journal of the American Medical Association. 2000;284(18).
- Transmissible Spongiform Encephalopathies in Humans Annu. Rev. Microbiol. 1999;53:283–314.
- Creutzfeldt-Jakob Disease Associated with Cadaveric Dura Mater Grafts.MMWR. November 14, 1997;46(45):1066-1069.
- Creutzfeldt-Jakob Disease in the United States, 1979-1994: Using National Mortality Data to Assess the Possible Occurrence of Variant Cases.EID. October-December 1996;2(4):333-337.
- Surveillance for Creutzfeldt-Jakob Disease, MMWR. August 9, 1996;45(31):665-668.
- Global surveillance, diagnosis, and therapy of human transmissible spongiform encephalopathies: Report of a WHO consultation, February 9-11, 1998, Geneva, Switzerland
- Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009, 132; 2659-2668
- National CJD Research & Surveillance Unit. Protocol: Surveillance of CJD in the UK (Accessed 15 Aug 2018)